Есть ли нервы у молочных зубов? — Bitesize Pediatric Dentistry
Иногда, когда ребенок теряет молочный зуб, он выглядит полым. Поскольку может показаться, что молочные зубы не так сложны, как постоянные зубы, и их все равно заменят, такие вещи, как кариес у детей или травма молочного зуба, могут не показаться огромной проблемой. Многие наши пациенты задаются вопросом, есть ли у молочных зубов нервы? Продолжайте читать, чтобы узнать правду.
Дело в том, что молочные зубы не только выполняют ряд очень важных функций, в том числе сохраняют место для правильного прорезывания постоянных зубов, но и анатомия молочных зубов такая же, как и у постоянных зубов. Таким образом, кариес у детей может болеть и, если его не лечить, привести к более серьезным проблемам.
Да, у молочных зубов есть нервы! Когда нервы повреждены или раздражены, зубная боль у ребенка может возникнуть так же, как и у взрослого. Как правило, этот тип боли зубного нерва возникает из-за травмы или большой полости, которая распространилась на пульпу зуба, достигая нервов внутри.
Чтобы дать вам лучшее представление о детских зубах, команда детской стоматологии Bitesize подготовила урок анатомии молочных зубов. В этом посте мы ответим:
- Молочные зубы полые?
- Из каких частей состоит молочный зуб?
- Почему необходимо лечить такие проблемы, как кариес у детей?
Молочные зубы полые?
К тому времени, как молочный зуб попадает к Зубной фее, может показаться, что внутри него ничего нет. Но молочные зубы не полые. На самом деле, когда постоянный зуб готовится прорезаться, его коронка давит на молочный зуб над ним. Это приводит к медленному растворению корней и внутреннего содержимого, из-за чего молочные зубы расшатываются и выпадают сами по себе без вмешательства. Именно поэтому выпадение молочных зубов обычно безболезненно.
Из каких частей состоит молочный зуб?
Зуб состоит из четырех основных частей, будь то молочный или постоянный зуб (в Stanford Children’s Health есть хорошая диаграмма частей зуба, если вы хотите увидеть):
Эмаль Внешний Слой молочного зуба называется эмалью. Зубная эмаль – самый твердый материал в организме. Он защищает зуб от бактерий, кислот, тепла и холода, а также от физических воздействий, которые могли бы повредить жизненно важные ткани внутри зуба. Хотя эмаль молочных зубов немного тоньше эмали постоянных зубов, она также может противостоять кариесу. Однако, как только кариес появляется, более тонкая эмаль означает, что он может распространяться быстрее.
Зубная эмаль – самый твердый материал в организме. Он защищает зуб от бактерий, кислот, тепла и холода, а также от физических воздействий, которые могли бы повредить жизненно важные ткани внутри зуба. Хотя эмаль молочных зубов немного тоньше эмали постоянных зубов, она также может противостоять кариесу. Однако, как только кариес появляется, более тонкая эмаль означает, что он может распространяться быстрее.
Дентин составляет внутренний слой и является крупнейшим структурным компонентом зуба. Имеет микроскопические канальцы. Если эмаль стирается и дентин обнажается, эти канальцы позволяют горячей, холодной, кислой или сладкой пище и напиткам стимулировать нервные клетки, вызывая чувствительность зубов. Задача дентина — поддерживать эмаль, чтобы дети могли жевать без разрушения эмали. Он также окружает мягкие ткани в центре зуба, защищая их от бактерий и других вредных вторжений.
Пульпа Пульпа — это мягкая ткань внутри зуба, находящаяся в пульповой камере.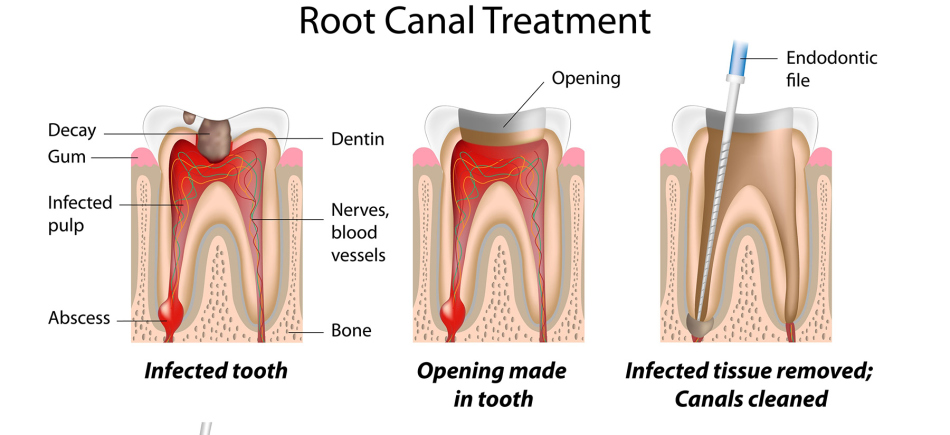 Он содержит нервы, соединительную ткань и кровеносные сосуды. Пульпа отвечает за кровоснабжение зуба, а ее сосудистая сеть соединяется с тканями вокруг зуба, включая периодонтальную связку. Пульпа молочного зуба может воспалиться, заразиться или даже отмереть, как и у взрослого зуба, поэтому вы можете лечить корневой канал молочного зуба, хотя эта процедура не так распространена, как у взрослых. .
Он содержит нервы, соединительную ткань и кровеносные сосуды. Пульпа отвечает за кровоснабжение зуба, а ее сосудистая сеть соединяется с тканями вокруг зуба, включая периодонтальную связку. Пульпа молочного зуба может воспалиться, заразиться или даже отмереть, как и у взрослого зуба, поэтому вы можете лечить корневой канал молочного зуба, хотя эта процедура не так распространена, как у взрослых. .
Корень зуба – это то, что удерживает зуб в кости челюсти. Он покрыт твердой соединительной тканью, известной как цемент, которая прикрепляет его к периодонтальной связке.
Почему нужно лечить такие проблемы, как кариес у детей?
Как видите, анатомия молочного зуба такая же, как и у постоянного зуба. Наши детские стоматологи в Парк-Слоуп, Вильямсбург и Дамбо, Бруклин лечат консервативно, и когда у ребенка возникает проблема, наша цель — остановить боль, максимально сохранить естественную структуру зуба и устранить любую инфекцию.
Если молочный зуб еще не готов к выпадению, но он разрушен или получил травму, идеальным решением будет сохранение зуба, чтобы он мог продолжать выполнять те важные функции, о которых мы упоминали. Если молочный зуб выпадает полностью или его необходимо удалить, часто устанавливается фиксатор зубного промежутка, чтобы другие зубы не смещались в оставшееся пространство и не вызывали скученности.
Если молочный зуб выпадает полностью или его необходимо удалить, часто устанавливается фиксатор зубного промежутка, чтобы другие зубы не смещались в оставшееся пространство и не вызывали скученности.
Посещение детского стоматолога как можно раньше, если ребенок получил травму или жалуется на зубную боль, или вы подозреваете, что у него есть кариес, повысит шансы на успешное лечение молочного зуба. Раннее обнаружение проблем также означает, что лечение будет менее инвазивным, что позволит нам избежать таких проблем, как корневые каналы молочных зубов.
Сохраним детские зубки крепкими и здоровыми!
Помимо здорового питания и тщательной чистки зубов щеткой и зубной нитью, регулярные визиты к детскому стоматологу будут иметь большое значение для предотвращения кариеса молочных зубов вашего ребенка. Если вы ищете веселого и дружелюбного детского дантиста в Бруклине, запишитесь на прием для своего малыша в детской стоматологии Bitesize уже сегодня!
Болезнь Шарко-Мари-Тута | Национальный институт неврологических расстройств и инсульта
Что такое болезнь Шарко-Мари-Тута?
Болезнь Шарко-Мари-Тута (ШМТ) относится к группе заболеваний, вызывающих поражение периферических нервов — нервов, передающих информацию и сигналы от головного и спинного мозга к остальным частям тела и от них, а также как сенсорная информация, такая как прикосновение, обратно в спинной и головной мозг. ШМТ также может напрямую воздействовать на нервы, контролирующие мышцы.
ШМТ также может напрямую воздействовать на нервы, контролирующие мышцы.
Прогрессирующая мышечная слабость обычно становится заметной в подростковом или раннем взрослом возрасте, но начало заболевания может возникнуть в любом возрасте. Поскольку в первую очередь поражаются более длинные нервы, симптомы обычно начинаются со стоп и голеней, а затем могут поражать пальцы, кисти и предплечья. Большинство людей с ШМТ имеют некоторую физическую инвалидность, хотя некоторые люди могут никогда не знать, что они больны.
ШМТ, также известная как наследственная моторная и сенсорная невропатия, является одним из наиболее распространенных наследственных неврологических заболеваний, от которого страдают примерно 126 000 человек в США и 2,6 миллиона человек во всем мире.
Почти все случаи передаются по наследству. Возможно наличие двух или более типов ШМТ, что происходит, когда у человека есть мутации в двух или более генах, каждый из которых вызывает определенную форму заболевания.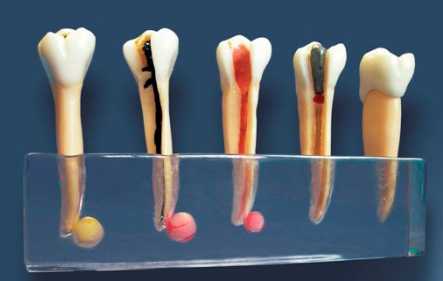 ШМТ является гетерогенным генетическим заболеванием, то есть мутации в разных генах могут вызывать сходные клинические симптомы. ШМТ названа в честь трех врачей, описавших ее в 1886 г.
ШМТ является гетерогенным генетическим заболеванием, то есть мутации в разных генах могут вызывать сходные клинические симптомы. ШМТ названа в честь трех врачей, описавших ее в 1886 г.
Симптомы
ШМТ поражает как чувствительные, так и двигательные нервы (нервы, которые запускают импульс для сокращения мышц) в руках, кистях, ногах и ступнях. Пораженные нервы медленно дегенерируют и теряют способность связываться со своими отдаленными целями, что приводит к мышечной слабости и атрофии в руках, ногах, кистях или ступнях.
Симптомы могут включать:
- Слабость или паралич мышц стопы и голени, которые могут вызывать затруднения при подъеме стопы (свисание стопы)
- Походка с высоким шагом с частыми спотыканиями или падениями
- Проблемы с балансом
- Деформации стопы, такие как высокий свод и искривление пальцев стопы (молоткообразные пальцы)
- Нижние конечности могут принять форму «перевернутой бутылки шампанского» из-за потери мышечной массы
- Снижение способности чувствовать тепло, холод и прикосновение
- Возможны слабость и атрофия кистей рук, вызывающие трудности с мелкой моторикой
- Снижение чувства вибрации и положения (проприоцепция)
- Искривление позвоночника (сколиоз)
- Смещение бедра
- Контрактуры (хроническое укорочение мышц или сухожилий вокруг суставов)
- Мышечные спазмы
- Нервная боль
Некоторым людям может потребоваться опора на стопы или опоры для ног или другие ортопедические приспособления для поддержания подвижности. Некоторые люди, живущие с ШМТ, испытывают тремор, а также могут быть затронуты зрение и слух. В редких случаях могут возникнуть затруднения дыхания, если поражены нервы, контролирующие мышцы диафрагмы.
Некоторые люди, живущие с ШМТ, испытывают тремор, а также могут быть затронуты зрение и слух. В редких случаях могут возникнуть затруднения дыхания, если поражены нервы, контролирующие мышцы диафрагмы.
Тяжесть симптомов может сильно различаться у разных людей и даже у членов семьи с заболеванием и генной мутацией. Прогрессирование симптомов постепенное.
Типы болезни Шарко-Мари-Тута
Существует множество различных типов ШМТ, которые могут иметь общие симптомы, но различаются по характеру наследования, возрасту начала заболевания и вовлечению аксона или миелиновой оболочки.
- CMT1 вызывается аномалиями миелиновой оболочки. Другие менее распространенные причины CMT1 возникают в результате мутаций в генах SIMPLE (также называемых LITAF), EGR2, PMP22 и NEFL соответственно.
- CMT1A является результатом дупликации гена на хромосоме 17, который несет инструкции по производству периферического миелинового белка-22 (PMP22). Белок PMP22 является критическим компонентом миелиновой оболочки.
 Сверхэкспрессия этого гена вызывает аномальную структуру и функцию миелиновой оболочки. CMT1A обычно медленно прогрессирует. С детства у людей наблюдается слабость и атрофия мышц голеней; позже они испытывают слабость в руках, потерю чувствительности и проблемы со стопами и ногами. Другая невропатия, отличная от CMT1A, называемая наследственной нейропатией с предрасположенностью к параличу от сдавления (HNPP), вызвана делецией одного из генов PMP22. В этом случае аномально низкие уровни гена PMP22 приводят к эпизодической рецидивирующей демиелинизирующей невропатии.
Сверхэкспрессия этого гена вызывает аномальную структуру и функцию миелиновой оболочки. CMT1A обычно медленно прогрессирует. С детства у людей наблюдается слабость и атрофия мышц голеней; позже они испытывают слабость в руках, потерю чувствительности и проблемы со стопами и ногами. Другая невропатия, отличная от CMT1A, называемая наследственной нейропатией с предрасположенностью к параличу от сдавления (HNPP), вызвана делецией одного из генов PMP22. В этом случае аномально низкие уровни гена PMP22 приводят к эпизодической рецидивирующей демиелинизирующей невропатии.
- CMT1A является результатом дупликации гена на хромосоме 17, который несет инструкции по производству периферического миелинового белка-22 (PMP22). Белок PMP22 является критическим компонентом миелиновой оболочки.
- CMT1B вызывается мутациями в гене, который несет инструкции по производству нулевого миелинового белка (MPZ, также называемого P0), который является еще одним важным компонентом миелиновой оболочки. Большинство этих мутаций являются точечными мутациями, то есть ошибка происходит только в одной букве генетического кода ДНК. На сегодняшний день ученые выявили более 120 различных точечных мутаций в гене P0. CMT1B вызывает симптомы, сходные с симптомами CMT1A.
- CMT2 является результатом аномалий аксона периферической нервной клетки, а не миелиновой оболочки, и встречается реже, чем CMT1. Это аутосомно-доминантное заболевание имеет более дюжины подтипов (некоторые из которых имеют свои варианты), причем каждый подтип связан с мутациями в определенном гене. Симптомы аналогичны симптомам, наблюдаемым при ШМТ1, но люди с ШМТ2 часто имеют меньшую инвалидность и потерю чувствительности, чем люди с ШМТ1. Начало ШМТ2 обычно приходится на детский или подростковый возраст. Некоторые типы ШМТ2 могут иметь поражение голосовых связок или диафрагмального нерва, вызывая проблемы с речью или дыханием.
- CMT3, или болезнь Дежерина-Сотта, представляет собой особенно тяжелую демиелинизирующую невропатию, которая начинается в младенчестве. Симптомы могут прогрессировать до тяжелой инвалидности, потери чувствительности и искривления позвоночника. Это редкое заболевание может быть вызвано мутациями в нескольких генах, включая PMP22, MPZ и EGR2, и может наследоваться либо доминантно, либо рецессивно. У младенцев:
- Тяжелая мышечная атрофия
- Слабость
- Задержка развития двигательных навыков
- Сенсорные проблемы
- CMT4 включает несколько различных подтипов демиелинизирующих, аксональных и моторных невропатий, которые наследуются аутосомно-рецессивно. Каждый подтип невропатии вызывается мутацией в другом гене (несколько генов были идентифицированы в CMT4). Мутации могут затрагивать определенную этническую популяцию и приводить к различным физиологическим или клиническим характеристикам. У людей с ШМТ4 симптомы слабости в ногах обычно появляются в детстве, а в подростковом возрасте они могут быть не в состоянии ходить. CMT4 редко встречается в США
- CMTX1 (также называемый CMT X, тип 1) — вторая по распространенности форма CMT. Это сцепленное с Х-хромосомой заболевание вызывается мутациями в гене, который обеспечивает инструкции по производству белка коннексина-32. Белок коннексин-32 обнаружен в миелинизирующих шванновских клетках — клетках, которые обвивают аксоны нервов и составляют миелиновую оболочку. У мужчин, унаследовавших мутантный ген, симптомы заболевания проявляются от умеренных до тяжелых, начиная с позднего детства или в подростковом возрасте. Женщины, которые наследуют мутировавший ген, часто имеют более легкие симптомы, чем мужчины, или вообще не проявляют симптомов.
 Сверхэкспрессия этого гена вызывает аномальную структуру и функцию миелиновой оболочки. CMT1A обычно медленно прогрессирует. С детства у людей наблюдается слабость и атрофия мышц голеней; позже они испытывают слабость в руках, потерю чувствительности и проблемы со стопами и ногами. Другая невропатия, отличная от CMT1A, называемая наследственной нейропатией с предрасположенностью к параличу от сдавления (HNPP), вызвана делецией одного из генов PMP22. В этом случае аномально низкие уровни гена PMP22 приводят к эпизодической рецидивирующей демиелинизирующей невропатии.
Сверхэкспрессия этого гена вызывает аномальную структуру и функцию миелиновой оболочки. CMT1A обычно медленно прогрессирует. С детства у людей наблюдается слабость и атрофия мышц голеней; позже они испытывают слабость в руках, потерю чувствительности и проблемы со стопами и ногами. Другая невропатия, отличная от CMT1A, называемая наследственной нейропатией с предрасположенностью к параличу от сдавления (HNPP), вызвана делецией одного из генов PMP22. В этом случае аномально низкие уровни гена PMP22 приводят к эпизодической рецидивирующей демиелинизирующей невропатии. CMT1B вызывает симптомы, сходные с симптомами CMT1A.
CMT1B вызывает симптомы, сходные с симптомами CMT1A. У младенцев:
У младенцев: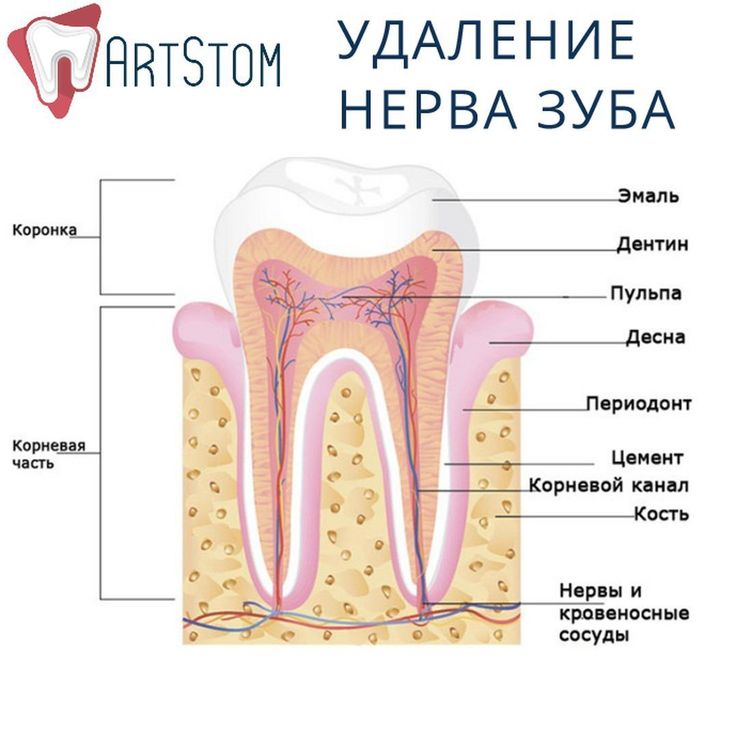 У мужчин, унаследовавших мутантный ген, симптомы заболевания проявляются от умеренных до тяжелых, начиная с позднего детства или в подростковом возрасте. Женщины, которые наследуют мутировавший ген, часто имеют более легкие симптомы, чем мужчины, или вообще не проявляют симптомов.
У мужчин, унаследовавших мутантный ген, симптомы заболевания проявляются от умеренных до тяжелых, начиная с позднего детства или в подростковом возрасте. Женщины, которые наследуют мутировавший ген, часто имеют более легкие симптомы, чем мужчины, или вообще не проявляют симптомов.У кого больше шансов заболеть болезнью Шарко-Мари-Тута?
ШМТ вызывается мутациями в генах, которые поддерживают или продуцируют белки, участвующие в структуре и функции либо аксона периферического нерва, либо миелиновой оболочки.
Нервная клетка передает информацию отдаленным целям, посылая электрические сигналы по длинной тонкой части клетки, называемой аксоном. Аксон окружен миелином, оболочкой, которая действует как изоляция на электрическом проводе и способствует высокоскоростной передаче электрических сигналов. Без интактного аксона и миелиновой оболочки сигналы, проходящие по нерву и аксону, либо медленны, либо имеют слабый сигнал, а это означает, что периферические нервные клетки становятся неспособными активировать мышцы или передавать сенсорную информацию от конечностей обратно в спинной мозг и спинной мозг. мозг.
мозг.
При ШМТ идентифицировано более 40 генов, каждый из которых связан с одним или несколькими типами заболевания. Кроме того, несколько генов могут быть связаны с одним типом CMT . Более половины всех случаев ШМТ вызваны дупликацией гена PMP22 на 17-й хромосоме.
Несмотря на то, что разные белки являются аномальными при различных формах ШМТ, все мутации в основном влияют на нормальную функцию периферических нервов. Генные дефекты миелина вызывают дисфункцию оболочки, которая искажает или блокирует нервные сигналы, в то время как другие мутации ограничивают функцию аксонов и вызывают их потерю.
Как наследуется болезнь Шарко-Мари-Тута
Генные мутации при ШМТ наследуются по трем различным схемам: аутосомно-доминантной, аутосомно-рецессивной и Х-сцепленной, причем все они связаны с хромосомами человека. У каждого человека 23 пары хромосом. Первые 22 пары называются «аутосомами» и наследуются независимо от биологического пола человека. Каждый человек обычно обладает двумя копиями каждого гена на аутосомах, по одной унаследованной от каждого родителя. Аутосомно-доминантный означает, что для развития заболевания необходима только одна копия гена ШМТ — от любого из родителей, а ребенок больного родителя имеет 50-процентный шанс унаследовать заболевание. Аутосомно-рецессивные заболевания возникают, когда ребенок получает два мутировавших гена, по одному от каждого родителя; ни один из родителей обычно не болел бы этим заболеванием. Их дети имеют 25-процентный шанс унаследовать болезнь. Аутосомные заболевания, как доминантные, так и рецессивные, в равной степени поражают мужчин и женщин.
Каждый человек обычно обладает двумя копиями каждого гена на аутосомах, по одной унаследованной от каждого родителя. Аутосомно-доминантный означает, что для развития заболевания необходима только одна копия гена ШМТ — от любого из родителей, а ребенок больного родителя имеет 50-процентный шанс унаследовать заболевание. Аутосомно-рецессивные заболевания возникают, когда ребенок получает два мутировавших гена, по одному от каждого родителя; ни один из родителей обычно не болел бы этим заболеванием. Их дети имеют 25-процентный шанс унаследовать болезнь. Аутосомные заболевания, как доминантные, так и рецессивные, в равной степени поражают мужчин и женщин.
Другие типы ШМТ наследуются сцепленно с Х-хромосомой, то есть зависят от хромосом, определяющих пол человека. У самок есть две Х-хромосомы, по одной унаследованной от каждого родителя. У мужчин есть X- и Y-хромосомы, причем Y-хромосома наследуется от родителя-мужчины. Ребенок мужского пола от матери, которая является носителем заболевания на одной из ее Х-хромосом, имеет 50-процентный шанс унаследовать это заболевание.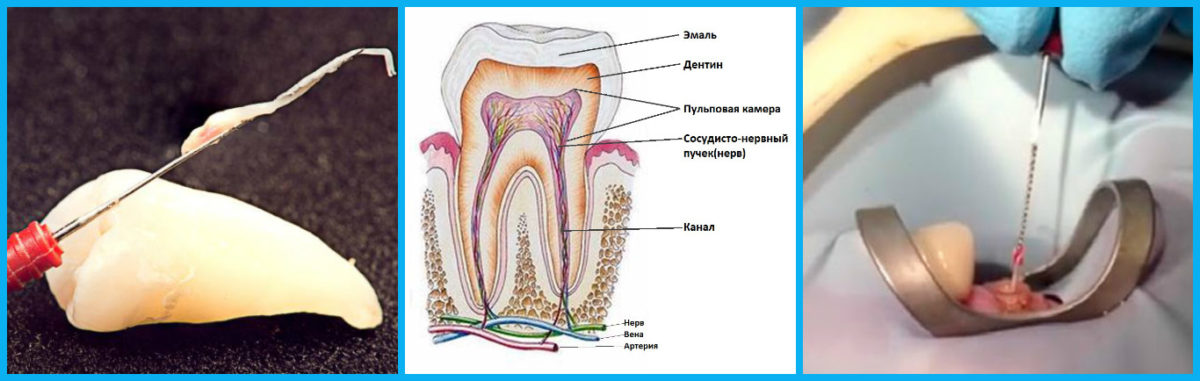
В некоторых случаях новая мутация возникает спонтанно в генетическом материале человека во время зачатия, без передачи по наследству в семье. Затем новая мутация может быть передана детям человека.
Как диагностируется и лечится болезнь Шарко-Мари-Тута?
Диагностика ШМТ
Диагностика начинается с подробного сбора анамнеза, семейного анамнеза и неврологического обследования. Врач будет искать признаки:
- Мышечная слабость в руках, ногах, кистях и стопах
- Снижение мышечной массы
- Сухожильные рефлексы снижены
- Сенсорная потеря
- Проблемы со стопой и ортопедические заболевания, такие как легкий сколиоз или аномальное формирование тазобедренного сустава
Специфическим признаком, который может быть обнаружен у пациентов с ШМТ1, является увеличение нерва, которое можно почувствовать или даже увидеть через кожу, особенно в области локтя. Эти увеличенные нервы, называемые гипертрофическими нервами, вызваны утолщением миелиновых оболочек.
Врач может заказать дополнительные исследования, в том числе:
- Исследования нервной проводимости
- Электромиография (ЭМГ)
- Генетическое тестирование
- Биопсия нерва
У людей с ШМТ1 обычно проявляются признаки аномальной миелинизации. В частности, можно увидеть образования, похожие на луковицы луковиц, которые представляют собой аксоны, окруженные слоями ремиелинизирующих шванновских клеток. У людей с ШМТ2 обычно наблюдаются признаки дегенерации аксонов без признаков демиелинизации.
Лечение ШМТ
ШМТ неизлечима, но физиотерапия и трудотерапия, корсеты и другие ортопедические приспособления, а также ортопедическая хирургия могут помочь при инвалидизирующих симптомах заболевания. Кроме того, при сильной нервной боли могут быть назначены обезболивающие препараты.
Важно поддерживать подвижность, гибкость и мышечную силу. Раннее начало программы лечения может отсрочить или уменьшить дегенерацию нервов и мышечную слабость до того, как они прогрессируют до инвалидности.
Туфли или сапоги с высоким голенищем также могут поддерживать слабые лодыжки. Шины для большого пальца могут помочь при слабости рук и потере мелкой моторики. Вспомогательные устройства следует использовать до того, как наступит инвалидность, поскольку они могут предотвратить мышечное напряжение и уменьшить мышечную слабость.
Какие последние новости о болезни Шарко-Мари-Тута?
Текущие исследования ШМТ включают усилия по выявлению большего количества мутантных генов и белков, вызывающих различные подтипы заболеваний, обнаружению механизмов дегенерации нервов и мышечной атрофии с целью разработки вмешательств для остановки или замедления этих изнурительных процессов и развития методы лечения дегенерации нервов и мышечной атрофии.
NINDS поддерживает Сеть клинических исследований NIH по редким заболеваниям, которая состоит из различных исследовательских консорциумов, целью которых является улучшение доступности информации о редких заболеваниях, клинических исследований и информации о клинических исследованиях. Консорциум сети по наследственным невропатиям проводит исследования, которые включают анализ естественного течения ШМТ, поиск новых генов и генов, которые изменяют симптомы человека, разработку терапии и обучающие программы для обучения будущих исследователей наследственных невропатий.
Консорциум сети по наследственным невропатиям проводит исследования, которые включают анализ естественного течения ШМТ, поиск новых генов и генов, которые изменяют симптомы человека, разработку терапии и обучающие программы для обучения будущих исследователей наследственных невропатий.
Ученые изучают регуляцию гена PMP22 для разработки и проверки тестов, которые измеряют присутствие, количество или активность целевого объекта. В других исследованиях изучается влияние малых молекул на биологическую систему с целью разработки новых методов лечения. Высокопроизводительные скрининги (способ быстрой оценки биологической активности большого количества соединений) могут выявить лекарства-кандидаты, снижающие уровень PMP22. Дополнительные исследования сосредоточены на том, как митохондрии, электростанция клетки, могут играть роль в дегенерации аксонов, наблюдаемой при ШМТ, а также при других заболеваниях.
Долговременное совместное исследование NIH надеется определить естественное течение ШМТ и то, как наличие определенной мутации гена может привести к типам и симптомам заболевания.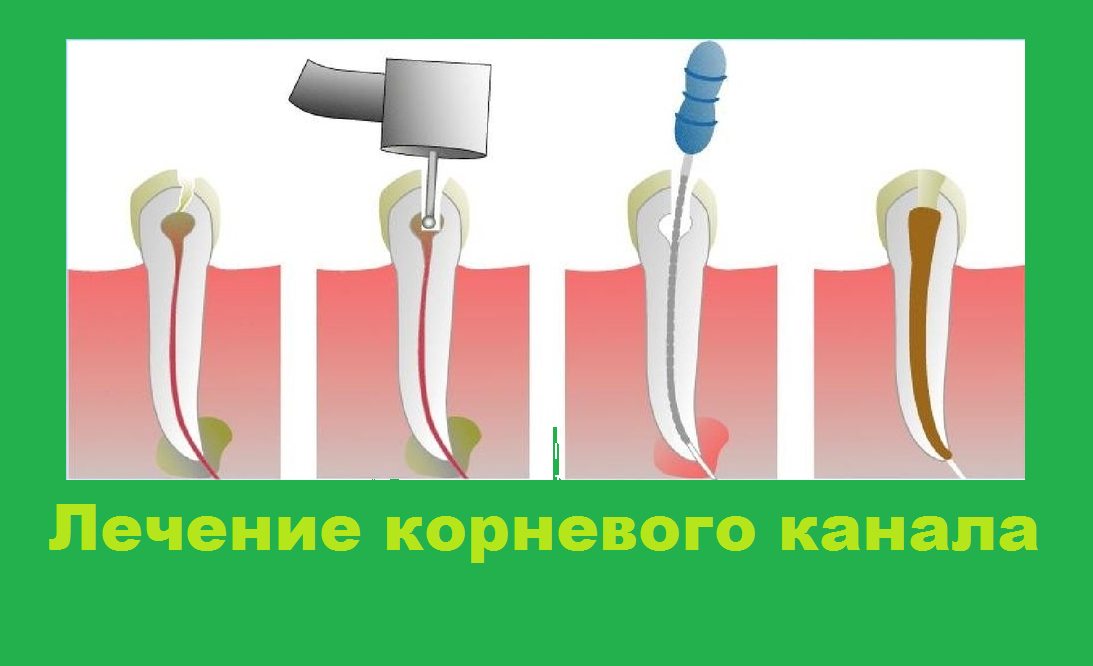 Кроме того, исследование, состоящее из двух частей, ищет новые гены, вызывающие заболевание, а также гены, которые не вызывают заболевание, но могут изменять симптомы человека. Другие ученые, финансируемые NIH, используют секвенирование следующего поколения (которое может одновременно быстро идентифицировать структуру миллионов небольших фрагментов ДНК) для идентификации новых генов CMT.
Кроме того, исследование, состоящее из двух частей, ищет новые гены, вызывающие заболевание, а также гены, которые не вызывают заболевание, но могут изменять симптомы человека. Другие ученые, финансируемые NIH, используют секвенирование следующего поколения (которое может одновременно быстро идентифицировать структуру миллионов небольших фрагментов ДНК) для идентификации новых генов CMT.
Генная терапия — еще одно многообещающее направление исследований. Эксперименты с использованием клеточных культур и моделей болезни на животных показали, что можно доставлять гены в шванновские клетки и мышцы. Другие исследования показывают, что трофические факторы или факторы роста нервов, такие как гормон андроген, предотвращают дегенерацию нервов.
file-medical
Узнайте о клинических испытаниях
Клинические испытания — это исследования, которые позволяют нам больше узнать о заболеваниях и улучшить лечение. Они могут помочь пациентам узнать о новых и предстоящих вариантах лечения.
Как я или мой близкий человек могу помочь улучшить уход за людьми с болезнью Шарко-Мари-Тута?
Рассмотрите возможность участия в клинических испытаниях, чтобы клиницисты и ученые могли больше узнать о ШМТ и связанных с ней расстройствах. В клинических исследованиях участвуют люди-добровольцы, чтобы помочь исследователям узнать больше о расстройстве и, возможно, найти более эффективные способы безопасного выявления, лечения или предотвращения болезни.
Нужны все типы добровольцев — здоровые или могут иметь заболевание или заболевание — всех возрастов, полов, рас и этнических групп, чтобы гарантировать, что результаты исследования применимы к как можно большему количеству людей, и что лечение будет безопасны и эффективны для всех, кто будет их использовать.
Для получения информации об участии в клинических исследованиях посетите сайт NIH Clinical Research Trials and You. Узнайте о клинических испытаниях, в которых в настоящее время ищут людей с ШМТ, на сайте Clinicaltrials.